3 Week 2- Working with Files
We will be following some of the data carpentry tutorial again (Copyright 2016 @ Software Carpentry) “Introduction to the command line for genomics”. We have made some modifications to the data carpentry tutorial to fit our course.
3.1 Learning Objectives
- Use shell commands and navigational shortcuts to efficiently navigate between files and folders
- Choose appropriate tools to view and search file contents
- Describe the general sequencing pipeline
3.2 Warm Up: Find the secret message
Start a new session in FARM OnDemand (refer to instructions here).
Your job is to figure out a secret phrase by seeing what letters you need to type
to access the folder genomes/mice/species/rudinoris/ultraprocessed/essential, starting from the directory /group/rbaygrp/eve198-genomics/week2_warmup/.
Use tab complete to find what letters you actually need to type to navigate to the folder folders. Write down those letters to form a secret phrase.
Once you make it to the essential directory, feel free to see what’s there!
Click for a hint
You can change directories with cd and you can view what is in each directory with ls
3.3 Viewing file contents
Now that you’ve made it to the essential folder, we have a few more tricks and commands you can learn!
From within the essential folder you, can see that there is a file called you_rule_too.txt
There are a few different ways to view the contents of that folder.
cat prints the context of the file into the terminal window.
$ cat you_rule_too.txtIf you have short files or they have contexts that you want to view along side
some of your recent terminal commands, cat is a great option. But if you have a
larger file, it might clog up your terminal window with text and make it hard to
see the commands you recently ran.
With a larger file, like bee_movie_full_script.txt, you might find that you
only want to view the starting or ending lines of the file.
Try running:
$ head bee_movie_full_script.txtAlso try:
$ tail bee_movie_full_script.txtNotice that head prints the first 10 lines of the file into the terminal, while
tail prints the last 10 lines of the file into the terminal.
If you want to be able to see the whole file, less opens a new window so you can
view and scroll through your file in the same way that man does. less also
works with some navigational tools such as:
| key | action |
|---|---|
| Space | to go forward |
| b | to go backward |
| g | to go to the beginning |
| G | to go to the end |
| q | to quit |
less also gives you a way of searching through files. Use the
“/” key to begin a search. Enter the word you would like
to search for and press enter to see the first occurance of that word.
Press “n”, and the screen will jump to the next location where that word is found.
Class Exercise 1
Use the navigation commands in
lessto explore the filebee_movie_full_script.txtOpen the file and try:
1. Go to the end of the file.
2. Go to the beginning of the file.
3. Find all the times that the script contains the phrase “dog”.
4. Go back to the end of the file. Try again to search of the phrase “dog”.
3.5 Sequencing Pipeline
To give you some context on the data we are working with today let’s talk about sequencing! It is important to understand the Illumina sequencing pipeline that produced the fastq files we will be working with. This video from Illumina explains the steps well, and I highly recommend checking it out: https://youtu.be/fCd6B5HRaZ8?si=68ZqTO2p2wRX-tUW. The sequencing workflow involves library preparation, cluster generation, sequencing and data analysis. We won’t get into the details of library preparation and cluster generation today, but the video does a good job of explaining it!
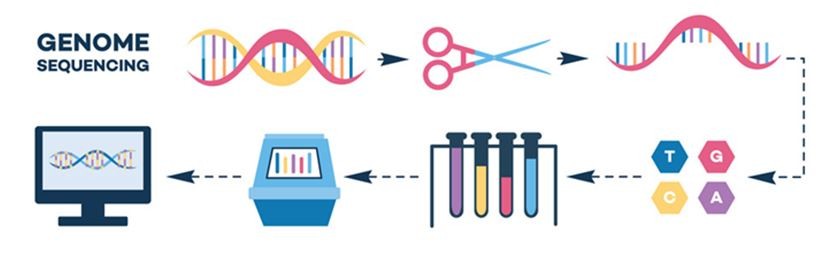

Below is also a figure highlighting the different steps of illumina sequencing, starting with the randomly fragmented genomic DNA and ending with sequence data! The figure is from this paper if you want to see more: https://www.sciencedirect.com/science/article/pii/S1359644608001244#fig3
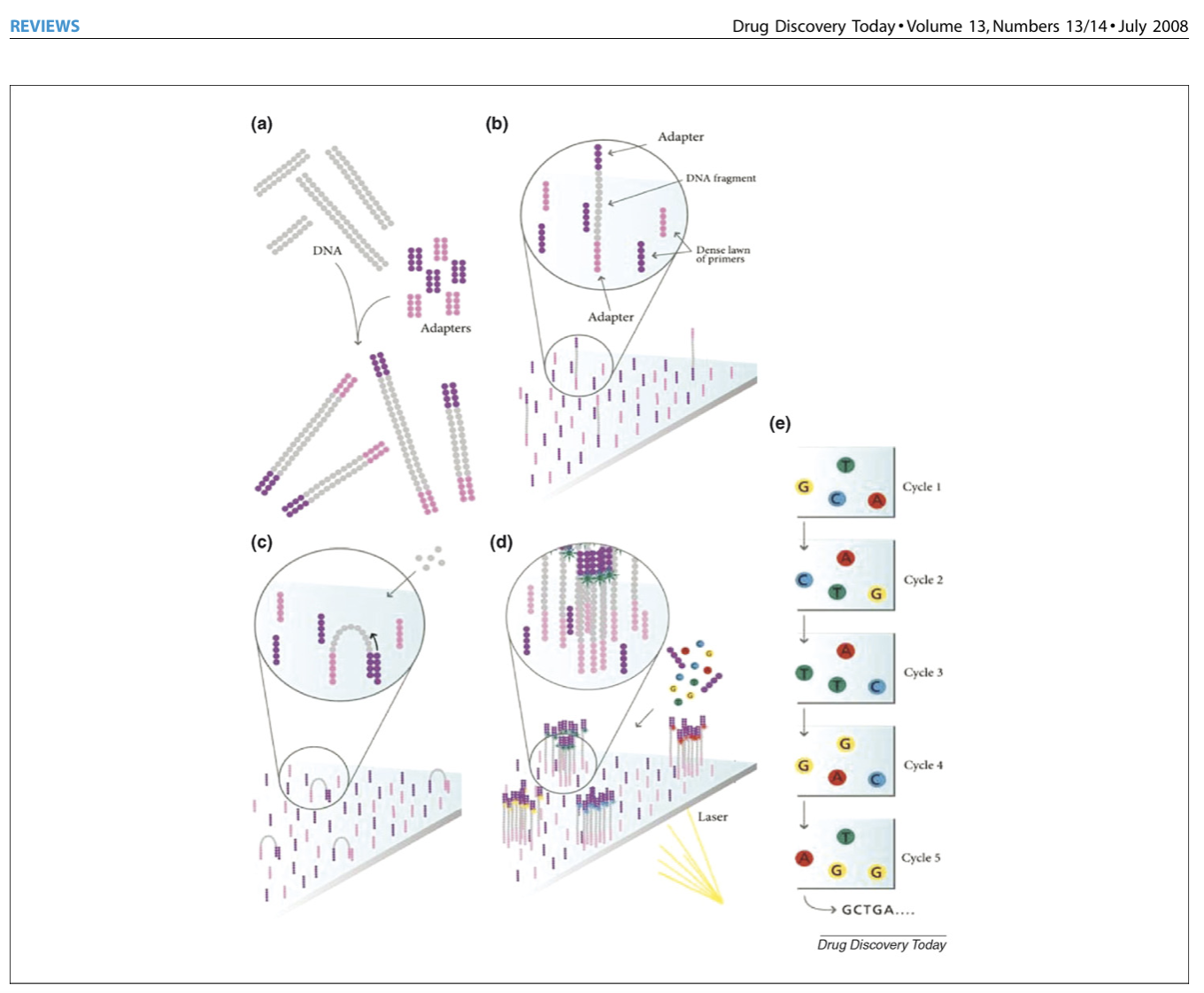 After we get our sequencing data back in .fq format, we can then use the package “fastqc” to process that data to fastq files which will help us understand the quality of of sequences. Now let’s talk about those files!
After we get our sequencing data back in .fq format, we can then use the package “fastqc” to process that data to fastq files which will help us understand the quality of of sequences. Now let’s talk about those files!
3.6 Details on the FASTQ format
A fastq file is a text file with information on the quality of the sequence data
Although it looks complicated (and it is), it’s easy to understand the fastq format with a little decoding. Some rules about the format include…
| Line | Description |
|---|---|
| 1 | Always begins with ‘@’ and then information about the read |
| 2 | The actual DNA sequence |
| 3 | Always begins with a ‘+’ and sometimes the same info in line 1 |
| 4 | Has a string of characters which represent the quality (phred) scores; must have same number of characters as line 2 |
With a full dataset you will also get an html file for each sample that give you more visual representations of your sample quality. These screenshots of a good and bad QC come from tutorials from the bioinformatics core linked here: https://bioinfo-core.org/index.php/9th_Discussion-28_October_2010 We will not have these visuals for our dataset today, but it is helpful to know what else you’d be working with if this was your own dataset!
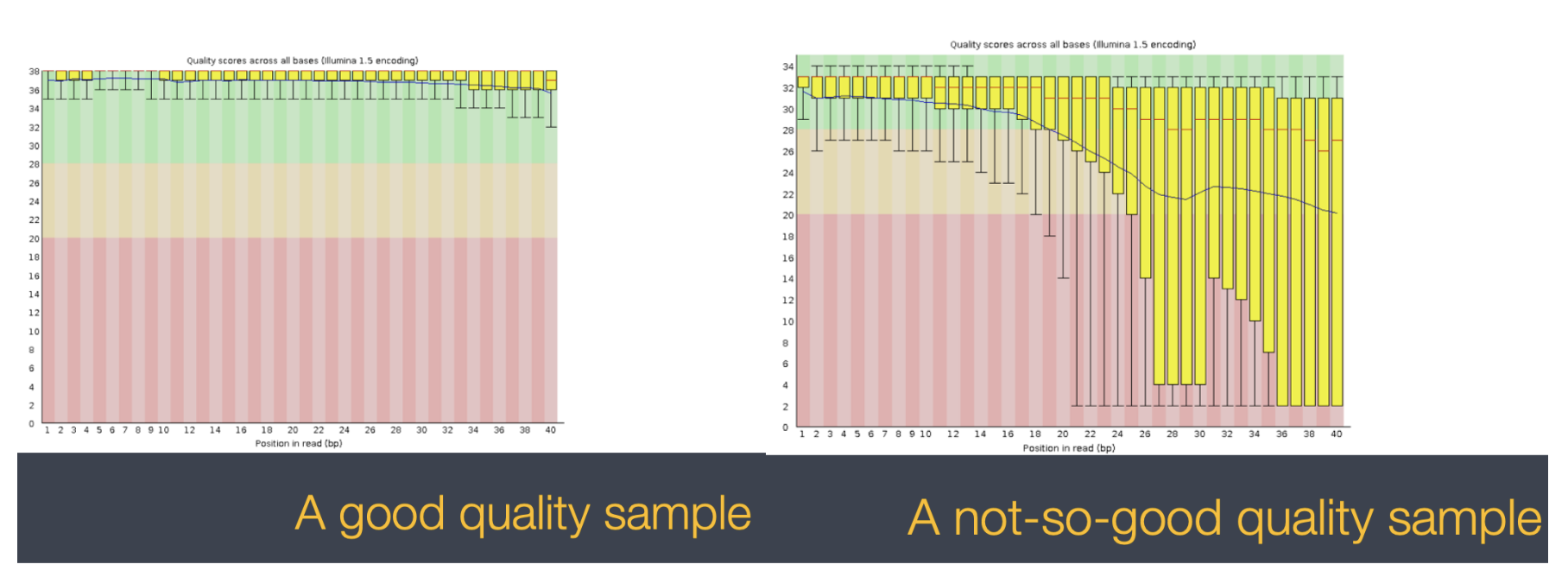
3.7 Our data set: FASTQ files
We can use the command wget which needs a link to the file that we want to download. If there’s a file saved on a website somewhere (anywhere on the internet) wget will download it for you. If our data file is on github, which is where most of our data will be stored, we’ll use the command git-clone
In this example we’re going to download all the material in our individual directories. Make sure you are in your personal directory, then type the following command:
$ wget https://raw.githubusercontent.com/mlarmstrong/IntroGenomics_Data/main/week2.tar.gzUse the tar command to uncompress the file. This will also automatically make a week2 directory in your individual directory:
tar -xzvf week2.tar.gzCheck out what is in this directory with the ls command. Remember you can do ls -l to see more information on the contents
untrimmed_fastq sra_metadata TableS2_QTL_Bay_2017.txtNow that we know how to navigate around our directory structure, let’s start working with our sequencing files. We did a sequencing experiment and have two results files, which are stored in our untrimmed_fastq directory.
3.8 Wildcards and echo
Navigate to your untrimmed_fastq directory in your week2 individual directory
$ cd /group/rbaygrp/eve198-genomics/yourdirectory/Week2/untrimmed_fastqWe are interested in looking at the FASTQ files in this directory. We can list all files with the .fastq extension using the command:
$ ls *.fastqSRR097977.fastq SRR098026.fastqThe * character is a special type of character called a wildcard, which can be used to represent any number of any type of character.
Thus, *.fastq matches every file that ends with .fastq.
This command:
$ ls *977.fastqSRR097977.fastqlists only the file that ends with 977.fastq.
This command:
$ ls /usr/bin/*.sh/usr/bin/gettext.sh /usr/bin/nvidia-bug-report.sh /usr/bin/nvidia-sleep.sh /usr/bin/rescan-scsi-bus.shLists every file in /usr/bin that ends in the characters .sh.
Note that the output displays full paths to files, since
each result starts with /.
echo is a built-in shell command that writes its arguments, like a line of text to standard output.
The echo command can also be used with pattern matching characters, such as wildcard characters.
Here we will use the echo command to see how the wildcard character is interpreted by the shell.
$ echo *.fastqSRR097977.fastq SRR098026.fastqThe * is expanded to include any file that ends with .fastq. We can see that the output of
echo *.fastq is the same as that of ls *.fastq.
What would the output look like if the wildcard could not be matched? Compare the outputs of
echo *.missing and ls *.missing.
$ echo *.missing*.missing$ ls *.missingls: cannot access '*.missing': No such file or directoryClass Exercise 3
Do each of the following tasks from your current directory using a single
lscommand for each:
- List all of the files in
/usr/binthat start with the letter ‘c’.- List all of the files in
/usr/binthat contain the letter ‘a’.- List all of the files in
/usr/binthat end with the letter ‘o’.Bonus: List all of the files in
/usr/binthat contain the letter ‘a’ or the letter ‘c’.Hint: The bonus question requires a Unix wildcard that we haven’t talked about yet. Try searching the internet for information about Unix wildcards to find what you need to solve the bonus problem.
3.9 Command History
If you want to repeat a command that you’ve run recently, you can access previous commands using the up arrow on your keyboard to go back to the most recent command. Likewise, the down arrow takes you forward in the command history.
A few more useful shortcuts:
- Ctrl+C will cancel the command you are writing, and give you a fresh prompt.
- Ctrl+R will do a reverse-search through your command history. This is very useful.
- Ctrl+L or the
clearcommand will clear your screen.
You can also review your recent commands with the history command, by entering:
$ historyto see a numbered list of recent commands. You can reuse one of these commands directly by referring to the number of that command.
For example, if your history looked like this:
259 ls *
260 ls /usr/bin/*.sh
261 ls *R1*fastqthen you could repeat command #260 by entering:
$ !260Type ! (exclamation point) and then the number of the command from your history.
You will be glad you learned this when you need to re-run very complicated commands.
For more information on advanced usage of history, read section 9.3 of
Bash manual.
3.10 Examining Files
Let’s revisit the different ways that we can view the contents of files.
Remember that we’ve introduced less, cat, head and tail.
Class Exercise 4
- Recall that
SRR098026.fastqis a folder that contains sequencing reads for an entire genome! Chat with you neighbor about which of the above commands might be most useful for viewing its contents. Try the method you chose to view the contents of your file.- What is the last line of the file?
- From your home directory, and without changing directories, use one short command to print the contents of all of the files in the
/group/rbaygrp/eve198-genomics/yourdirectory/week2/untrimmed_fastqdirectory.- What are the next three nucleotides (characters) after the first instance of the sequence “ACA”?
Let’s use the head and tail to look at the beginning and end of one of our .fastq files.
$ head SRR098026.fastq@SRR098026.1 HWUSI-EAS1599_1:2:1:0:968 length=35
NNNNNNNNNNNNNNNNCNNNNNNNNNNNNNNNNNN
+SRR098026.1 HWUSI-EAS1599_1:2:1:0:968 length=35
!!!!!!!!!!!!!!!!#!!!!!!!!!!!!!!!!!!
@SRR098026.2 HWUSI-EAS1599_1:2:1:0:312 length=35
NNNNNNNNNNNNNNNNANNNNNNNNNNNNNNNNNN
+SRR098026.2 HWUSI-EAS1599_1:2:1:0:312 length=35
!!!!!!!!!!!!!!!!#!!!!!!!!!!!!!!!!!!
@SRR098026.3 HWUSI-EAS1599_1:2:1:0:570 length=35
NNNNNNNNNNNNNNNNANNNNNNNNNNNNNNNNNN$ tail SRR098026.fastq+SRR098026.247 HWUSI-EAS1599_1:2:1:2:1311 length=35
#!##!#################!!!!!!!######
@SRR098026.248 HWUSI-EAS1599_1:2:1:2:118 length=35
GNTGNGGTCATCATACGCGCCCNNNNNNNGGCATG
+SRR098026.248 HWUSI-EAS1599_1:2:1:2:118 length=35
B!;?!A=5922:##########!!!!!!!######
@SRR098026.249 HWUSI-EAS1599_1:2:1:2:1057 length=35
CNCTNTATGCGTACGGCAGTGANNNNNNNGGAGAT
+SRR098026.249 HWUSI-EAS1599_1:2:1:2:1057 length=35
A!@B!BBB@ABAB#########!!!!!!!######By default, head and tail show us 10 lines, but we can use the -n option
with either of these commands to print the first or last n lines of a file.
$ head -n 1 SRR098026.fastq@SRR098026.1 HWUSI-EAS1599_1:2:1:0:968 length=35$ tail -n 1 SRR098026.fastqA!@B!BBB@ABAB#########!!!!!!!######Recall that a .fastq file has 4 lines per sequencing read. We can view the first complete read in one of the files in our dataset by using head to look at the first four lines.
$ head -n 4 SRR098026.fastq@SRR098026.1 HWUSI-EAS1599_1:2:1:0:968 length=35
NNNNNNNNNNNNNNNNCNNNNNNNNNNNNNNNNNN
+SRR098026.1 HWUSI-EAS1599_1:2:1:0:968 length=35
!!!!!!!!!!!!!!!!#!!!!!!!!!!!!!!!!!!All but one of the nucleotides in this read are unknown (N). This is a pretty bad read!
Line 4 shows the quality for each nucleotide in the read. Quality is interpreted as the probability of an incorrect base call (e.g. 1 in 10) or, equivalently, the base call accuracy (e.g. 90%). To make it possible to line up each individual nucleotide with its quality score, the numerical score is converted into a code where each individual character represents the numerical quality score for an individual nucleotide. For example, in the line above, the quality score line is:
!!!!!!!!!!!!!!!!#!!!!!!!!!!!!!!!!!!The # character and each of the ! characters represent the encoded quality for an
individual nucleotide. The numerical value assigned to each of these characters depends on the
sequencing platform that generated the reads. The sequencing machine used to generate our data
uses the standard Sanger quality PHRED score encoding, Illumina version 1.8 onwards.
Here is a link showing what those different symbols mean for quality scores: https://help.basespace.illumina.com/files-used-by-basespace/quality-scores
Each character is assigned a quality score between 0 and 42 as shown in the chart below.
Quality encoding: !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJK
| | | | |
Quality score: 0........10........20........30........40.. Each quality score represents the probability that the corresponding nucleotide call is incorrect. This quality score is logarithmically based, so a quality score of 10 reflects a base call accuracy of 90%, but a quality score of 20 reflects a base call accuracy of 99%. These probability values are the results from the base calling algorithm and dependent on how much signal was captured for the base incorporation.
Looking back at our read:
@SRR098026.1 HWUSI-EAS1599_1:2:1:0:968 length=35
NNNNNNNNNNNNNNNNCNNNNNNNNNNNNNNNNNN
+SRR098026.1 HWUSI-EAS1599_1:2:1:0:968 length=35
!!!!!!!!!!!!!!!!#!!!!!!!!!!!!!!!!!!We can now see that the quality of each of the Ns is 0 and the quality of the only
nucleotide call (C) is also very poor (# = a quality score of 2). This is indeed a very
bad read.
3.11 Group Work Activity- Examining a Fastq File
Make a directory within your personal directory to work on the week 2 activity.
cd /group/rbaygrp/eve198-genomics/yourdirectory
mkdir week2_activity
cd week2_acitvityCopy the ‘CCGPMC004_M0D060025C_S150_L003_R1_001.fastq.gz’ file in the week2_activity directory to your individual directory. This is a large file so it might take a minute!
cp ../../week2_activity/CCGPMC004_M0D060025C_S150_L003_R1_001.fastq.gz ./Next you will need to unzip the fastq file. To do this we will use the command “gunzip”. This unzips a gzipped file. “gzip” zips a file again. It will take a second since this is a larger file than our previous examples. It is actually an urchin sample from a project that was published out of our lab! (See publication here)
gunzip CCGPMC004_M0D060025C_S150_L003_R1_001.fastq.gzThen answer the following questions and submit them to the “Week 2: Examining Files” assignment on canvas. I would not recommend using ‘less’ or ‘cat’ on this file due to its size.
- Write out the first two lines of CCGPMC004_M0D060025C_S150_L003_R1_001.fastq
- Remember that line 4 in a fastq file shows the quality (phred) scores. If you look at line 4 and reference the score table from the link and figures below. What is the most common symbol, Q-score and error probability for most of our nucleotide calls?
https://help.basespace.illumina.com/files-used-by-basespace/quality-scores
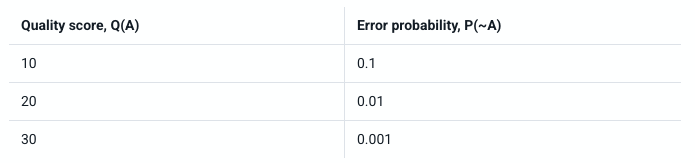
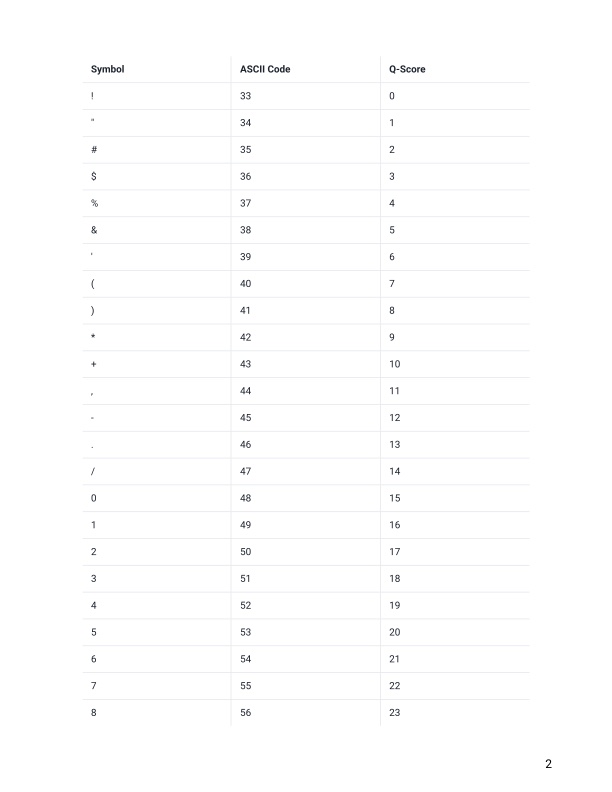
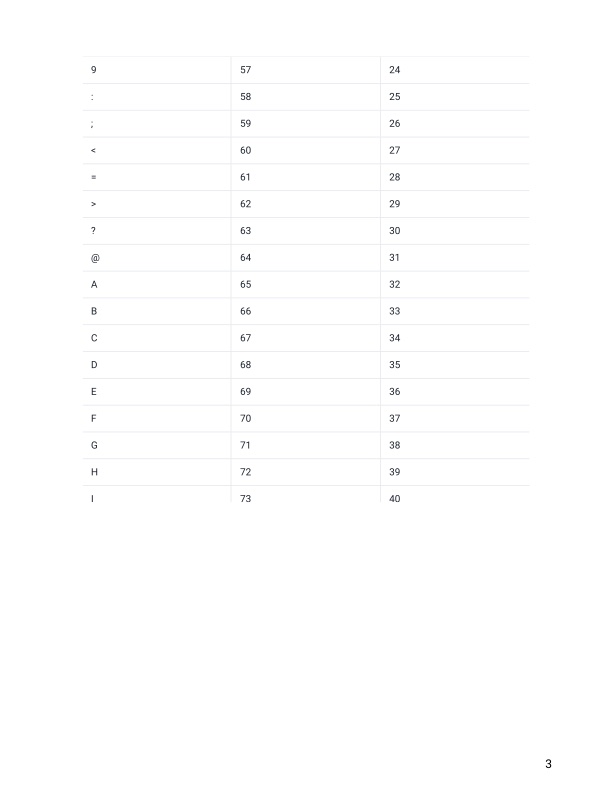
3.12 Key Points
The /, ~, and * characters represent important navigational shortcuts.
The
historycommand and the up arrow on your keyboard can be used to repeat recently used commands.You can view file contents using
less,cat,headortail.Sequencing reads are stored in .fastq files which have 4 lines per read.
Class Exercise Solutions
Exercise 1: Solution
- After typing “less bee_movie_full_script.txt” type “G” to see ” ”
- Type “g” to see ”
- Type “/dog” and press “n”. Should find two occurances: ” ” and ” ”
- Type “G”, then if you retry “/dog” nothing comes up.
Exercise 3: Solution
ls /usr/bin/c*ls /usr/bin/*a*ls /usr/bin/*o
Bonus:ls /usr/bin/*[ac]*Exercise 4: Solution
catis probably the worst option. If you want to look at the start or end, you could useheadortail, respectively.lessis a great option if you’d like to scroll through the file or search for any specific phrases.- The last line of the file is
C:CCC::CCCCCCCC<8?6A:C28C<608'&&&,'$.cat ./~/group/rbaygrp/eve198-genomics/yourdirectory/week2/untrimmed_fastq/*- GCG”